Genome-Wide Resolution of Chromatin Interactions
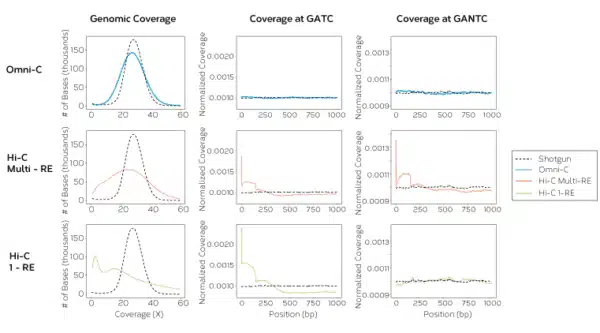
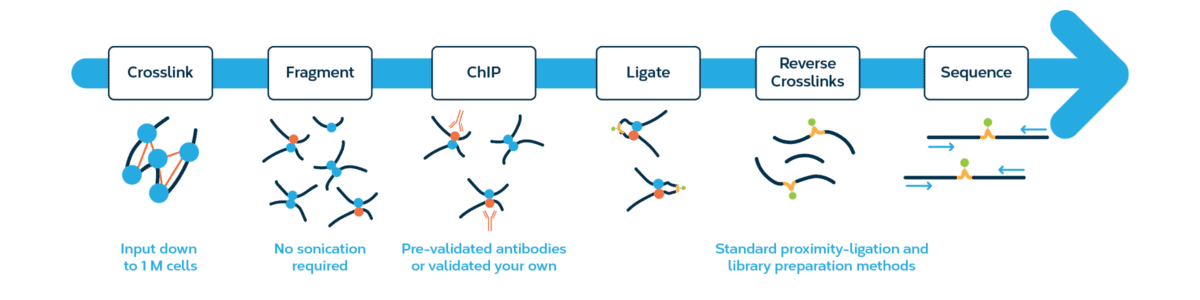
The Omni-C kit has optimized DNase I, a sequence-independent endonuclease, to reproducibly digest chromatin, delivering all the characteristics of a Hi-C approach without the sequence bias inherent to restriction enzyme-based Hi-C approaches.
Get enriched long-range cis reads, and more complete contact matrices when viewing chromatin conformation and looping interactions. Omni-C data gives you the most complete genome-wide view for all your genome assembly, phasing, structural variant, and SNP research.