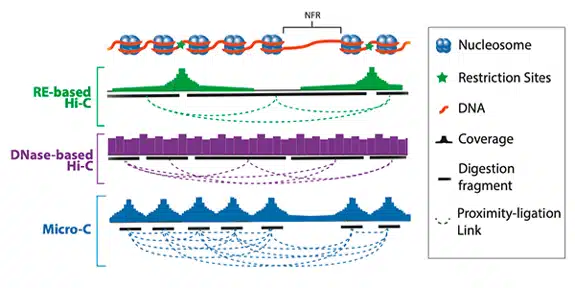
Customer Testimonials
“The Dovetail® Micro-C Kit transformed how I approach my research. It is enabling the analysis of distal chromatin interactions that are critical to developing a full understanding of human disease.”
Dr. Yasuhiro Murakawa - Team Leader, RIKEN-IFOM Joint Laboratory for Cancer Genomics and Professor - Kyoto University